Slide
Show JP
☰
探索
サインイン
サインアップ
アップロード
×
ダウンロード
カテゴリーなし
ソフトマターの物理学
Document
アンケートの質問への回答
両豊記による豊前
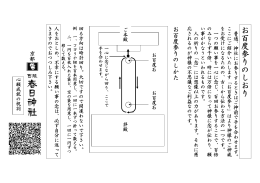
お 百 度 参 り の し お り
動 的 光 散 乱 装 置(DLS)
1998.9.14 DTL四極磁石の渦電流の評価