Slide
Show JP
☰
探索
サインイン
サインアップ
アップロード
×
ダウンロード
カテゴリーなし
たかがアセンブリ?―次世代シーケンス解析はじめの一歩―
論文の内容の要旨
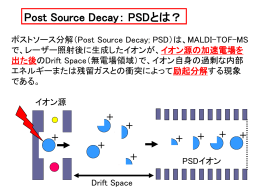
PSD+de novo sequence